經濟影響與成本效益 盡管初期投入較高(平均每企業需50萬歐元),但eCTD可減少30%的審評延遲成本,長期效益。仿制藥企業通過eCTD復用原研數據,節省80%的申報準備時間。歐盟預算撥款2億歐元資助中小企業完成數字化轉型。 倫理審查與數據隱私 eCTD中的患者數據需匿名化處理,符合《通用數據保護條例》(GDPR)要求。臨床試驗模塊(模塊5)的提交需附帶倫理委員會批準文件,且區域版本需體現各國倫理審查差異。AI輔助匿名化工具在保護隱私的同時提升數據處理效率。 技術融合與跨領域應用 eCTD格式擴展至醫療器械和保健品領域,歐盟試點eCTD-MDR項目整合ISO標準。基因產品的eCTD需附加生物安全數據庫,并與歐盟基因庫實時同步。未來,eCTD或與電子健康檔案(EHR)系統對接,支持個性化用藥。 持續改進與行業反饋機制 EMA每年發布eCTD實施報告,分析常見錯誤并更指南。行業聯盟(如EFPIA)通過定期研討會向監管機構反饋技術痛點,推動標準優化。開放式API接口的推廣將促進eCTD工具鏈的互操作性,降低技術鎖定風險。歐盟CESP提交通道相關技術支持。山東國產eCTD文件如何制作
技術壁壘與興市場挑戰 非洲和東南亞國家逐步采納eCTD,但其IT基礎設施薄弱導致實施進度滯后。歐盟通過“eCTD全球化倡議”提供技術援助,幫助興市場建立驗證體系和培訓中心。跨國藥企需針對不同區域定制遞交策略,例如在模塊1附加本地穩定性數據。 監管科學與創激勵 eCTD支持真實世界證據(RWE)和適應性臨床試驗設計的整合,加速創藥上市。EMA的PRIME計劃為突破性療法提供eCTD快速通道,允許分階段提交模塊數據。孤兒藥和兒科藥的eCTD序列可享受費用減免和優先審評。 供應鏈安全與審計追蹤 eCTD的XML主干文件記錄所有提交版本,支持供應鏈問題的追溯分析。原料藥CEP持有者需及時更變更信息,確保下游制劑廠商獲取數據。區塊鏈技術試點用于追蹤eCTD數據流,防止篡改和未授權訪問。 文化差異與實施障礙 部分南歐國家偏好傳統紙質流程,導致eCTD推廣阻力較大。EMA通過多語種培訓材料和區域協調員制度促進文化適應。行業需調整管理思維,將eCTD從“合規負擔”轉化為“競爭優勢”。山東國產eCTD文件如何制作加拿大eCTD申報相關技術支持。

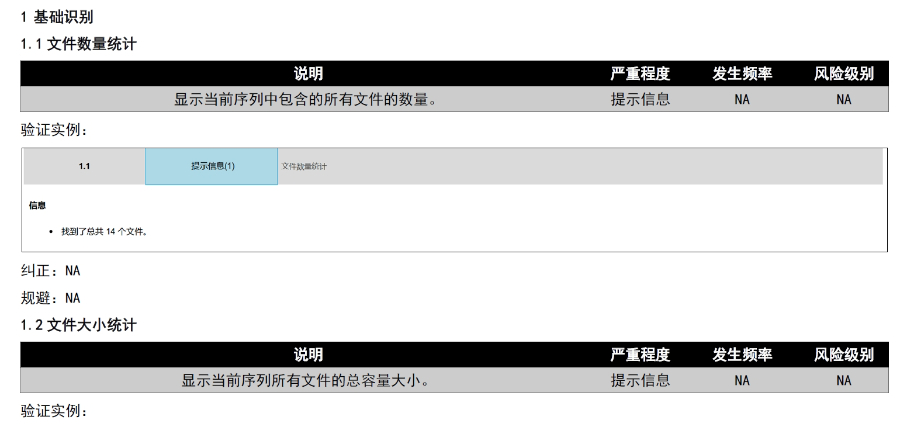
eCTD文件制作需遵循嚴格的法規要求和標準化流程,以下是關鍵要點整理:eCTD采用模塊化結構,包含模塊1(行政信息)至模塊5(臨床報告),需按ICH和監機構要求構建目錄樹。顆粒度選擇:文件提交層級需在***申報時確定并沿用,例如原料和制劑的章節(如、)需按比較低顆粒度拆分,輔料單獨成章。PDF需添加書簽(導航目錄)和超鏈接(跨網頁跳轉),超過5頁的文件必須包含目錄(TOC/LOT/LOF)。技術參數:初始視圖需設置默認縮放級別和頁面布局,書簽展開層級不超過三級,單文件大小需符合申報系統限制。驗證工具:使用軟件(如BXeCTD)自動生成書簽和超鏈接,并通過序列校驗和PDF校驗功能確保合規性。
文件生命周期管理:eCTD支持文件替換(Replace)、刪除(Delete)等操作,而非增文件。例如,更臨床研究方案時需用Replace操作覆蓋舊版本。基線提交(Baseline Submission)可用于補充歷史紙質資料,但需在封面函中聲明無內容變更。 臨床數據與研究標簽文件(STF):模塊4和5中的研究數據需通過STF(Study Tagging Files)引用,確保數據與文檔關聯。FDA要求數據集(如SAS XPORT格式)能置于模塊3-5,且單個文件超過4GB需拆分。2022年統計顯示,58%的ANDA因研究數據技術拒絕標準(TRC)錯誤被拒。 電子簽名與表格要求:FDA表格(如356h、1571)需使用數字簽名,PDF文件禁止加密或設置編輯限制。電子簽名需符合21 CFR Part 11規范,確保身份驗證、不可否認性和數據完整性。 外包服務與系統解決方案:賦悅科技累計提交超2000份eCTD申請,外包可降低40%人工錯誤率。美國eCTD注冊外包相關技術支持。
美國電子提交通道ESG(Electronic Submissions Gateway)是美國食品藥品監督管理局(FDA)建立的電子化監管信息提交系統,旨在為制藥、生物制品、醫療器械等行業提供安全、高效的電子申報服務。自2006年啟用以來,ESG已成為FDA接收電子監管材料的入口,每日處理上千份提交文件,涵蓋上市前審批、上市后監管、臨床試驗數據、不良反應報告等多種類型。該系統通過數字證書加密和公鑰基礎設施(PKI)技術,確保文件傳輸的真實性、完整性和不可否認性,符合FDA對電子提交的嚴格合規要求。在技術層面,ESG具備強大的文件處理能力。2018年系統升級后,取消了單個文件8GB的限制,可支持高達35GB的大型文件提交,進一步滿足復雜申報需求。此外,文件格式需遵循eCTD(電子通用技術文檔)規范,包括模塊化結構、PDF標準化和XML元數據整合,以確保全球監管機構兼容性。2025年3月28日起,FDA將啟用新一代平臺ESG NextGen,逐步替代現有系統,過渡期需關注兼容性和穩定性問題。瑞士eCTD注冊外包相關技術支持。山東國產eCTD文件如何制作
美國注冊鄧白氏號申請相關技術支持。山東國產eCTD文件如何制作
eCTD生命周期管理與變更提交:歐盟要求eCTD申報資料覆蓋藥品全生命周期,包括提交、補充申請及實質性變更。例如,增成員國需提交“附加成員國序列”,審評時間約52-83天;重大變更(如生產工藝調整)需創建序列并通過CTIS平臺更模塊3和模塊1的GMP證明。技術驗證工具(如EDQM推薦的檢查軟件)需在每次提交前運行,確保XML骨架文件與PDF書簽層級符合規范。此外,電子簽章需符合《歐盟電子簽名法》,并在模塊1中明確標注法律效力。歐洲通用提交門戶(Common European Submission Portal,CESP)是歐盟及成員國藥品監管機構間用于電子化提交申報資料的重要平臺。以下是關于CESP的詳細介紹: CESP是由歐盟藥品監管部門負責人網絡(HMA)合作開發的在線交付系統,旨在為藥品注冊申請者、利益相關方和監管機構之間提供統一、安全的電子提交通道。其設計初衷是簡化跨國申報流程,允許通過單一門戶向多個歐洲國家的藥監部門同時提交申請,避免了重復操作。山東國產eCTD文件如何制作
賦悅科技(杭州)有限責任公司在同行業領域中,一直處在一個不斷銳意進取,不斷制造創新的市場高度,多年以來致力于發展富有創新價值理念的產品標準,在浙江省等地區的數碼、電腦中始終保持良好的商業口碑,成績讓我們喜悅,但不會讓我們止步,殘酷的市場磨煉了我們堅強不屈的意志,和諧溫馨的工作環境,富有營養的公司土壤滋養著我們不斷開拓創新,勇于進取的無限潛力,賦悅科技供應攜手大家一起走向共同輝煌的未來,回首過去,我們不會因為取得了一點點成績而沾沾自喜,相反的是面對競爭越來越激烈的市場氛圍,我們更要明確自己的不足,做好迎接新挑戰的準備,要不畏困難,激流勇進,以一個更嶄新的精神面貌迎接大家,共同走向輝煌回來!